
EHDN 2014 Barcellona: Cosa è emerso in termini di ricerca e prospettive di cura

Dove va la ricerca clinica e cosa dobbiamo attenderci
Editoriale a cura di Ferdinando Squitieri (06/10/2014)
Dagli ultimi congressi internazionali, incluso quello europeo di Barcellona che si è svolto dal 19 al 21 settembre scorsi, si evince chiaramente che stiamo entrando in una fase storica della ricerca sulla malattia di Huntington in cui le scoperte di laboratorio cominciano ad integrarsi con la ricerca clinica. Per la prima volta infatti sono numerose le sperimentazioni di nuovi farmaci che si proporranno nel panorama del prossimo futuro. Da una sconfortante fase di lunghi silenzi durante cui sembrava non esserci una luce alla fine del tunnel entriamo in un'altra in cui i tentativi di modificare la gravità della malattia sembrano essere numerosi e promettenti. La ricerca di base, quella sulle cellule, sui modelli animali, in altri termini quella dei laboratorio attrezzati a ricercare nuovi farmaci comincia a tradursi in concreta speranza per i pazienti. Cosa allora ci dobbiamo aspettare nel prossimo futuro, quali sono le reali prospettive e come il nostro Paese si pone nello scenario della ricerca internazionale. I fronti su cui la ricerca scientifica sembra concentrarsi per arrivare alla cura sono fondamentalmente tre:
1) Identificazione di biomarkers;
2) Sviluppo di nuove tecnologie terapeutiche;
3) Applicazione delle nuove tecnologie alle sperimentazioni farmacologiche con nuovi composti sviluppati in laboratorio.
Questo è ancora una volta emerso, in un modo ancora più convincente, dal congresso europeo sulla malattia di Huntington di Barcellona.
Identificazione di biomarkers
L'identificazione di biomarkers rappresenta una determinante fase preliminare rispetto alla cura. Se desideriamo prevenire la malattia in chi ha scoperto di avere una mutazione genetica, abbiamo bisogno di interpretare segnali biologici in grado di fornirci quelle informazioni sul processo patologico che si avvicina e determina i sintomi in loro assenza.
Tre importanti progetti stanno da tempo conducendo ricerche importanti a tale riguardo. Si chiamano TRACK HD, PREDICT e HD-MAPS. I primi due studiano modifiche del sistema nervoso rispetto ai cambiamenti clinici nei pazienti anche in fase presintomatica, il terzo, cui partecipa anche il gruppo di Neurogenetica dell’IRCSS Neuromed, ricerca fattori genetici in grado di modificare lo sviluppo della malattia. TRACK e PREDICT hanno individuato possibili marcatori/biomarkers che verranno presto utilizzati nelle prossime sperimentazioni farmacologiche, grazie all'applicazione di RMN cerebrale. È il caso della sperimentazione di prossimo inizio con un farmaco immunomodulatore che si spera sia in grado di rallentare il decorso della malattia. La sperimentazione si chiamerà LEGATO-HD, ci coinvolgerà per cui ci stiamo preparando ad affrontarla nella maniera migliore e sarà sostenuta economicamente da TEVA Pharma, l'industria Israeliana che è già attualmente impegnata con la sperimentazione con Pridopidina (ex Huntexil) tuttora in corso con il nostro impegno congiunto di Neuromed e LIRH in Italia. Prevediamo che LEGATO-HD abbia inizio tra fine anno e inizio del prossimo e vedrà, per la prima volta, includere pazienti molto poco sintomatici, quasi presintomatici. Questa è una grande novità. I due progetti TRACK e PREDICT sono condotti da personalità scientifiche di altissimo spessore come Sarah Tabrizi per l'Inghilterra, Jane Paulsen per gli USA ed il nostro Ralf Reilmann, membro del consiglio scientifico di LIRH, per la Germania. A margine di questi importanti progetti molto ben finanziati dalla fondazione americana CHDI, si articolano iniziative scientifiche altrettanto valide che contribuiscono a fare emergere nuove conoscenze sulla storia naturale della malattia. Il gruppo di Neurogenetica dell’IRCCS Neuromed collabora da anni col gruppo del Dr. Umberto Sabatini (IRCCS Santa Lucia di Roma, anche lui membro del comitato scientifico di LIRH) per individuare nuovi marcatori cerebrali attraverso l'analisi di tecnologie innovative di RMN (risonanza magnetica).
Gli studi che hanno evidenziato importanti novità sulle modifiche dell'accumulo di ferro in alcune strutture cerebrali ed alterazioni del corpo calloso, la più importante struttura di collegamento tra diverse parti del cervello, sono state apprezzatissime dal mondo scientifico e dagli organizzatori del congresso di Barcellona. Oggi siamo pertanto in grado di individuare sottili cambiamenti già prima della malattia e, sebbene queste metodiche siano applicabili ancora solo a progetti di ricerca, ci avviciniamo un po' alla volta all'uso pratico a scopo terapeutico.
Affascinante e promettente è la ricerca di fattori genetici in grado di modificare l'evoluzione della malattia di Huntington. La mutazione da espansione di triplette CAG rappresenta certamente la causa della malattia, ma sembra influenzarne presentazione e gravità solo in parte. Cosa altro incide su questi aspetti e quale rapporto esiste tra la genetica e l'ambiente in cui viviamo? Questi sono gli interrogativi che si pone il progetto HD-MAPS, coordinato e presentato a Barcellona da James Gusella, già principale scopritore del gene dell'Huntington, dell'Università di Harvard, Boston, Stati Uniti, alla solita lucida, chiara e brillante maniera. Il campione di sangue di tante famiglie seguite dal sottoscritto contribuisce a questo importante progetto. Oggi sappiamo che, per iniziare a trovare elementi scientifici significativi sono stati necessari almeno 1500 campioni biologici provenienti da ogni parte del mondo. Sono stati necessari 4000 campioni di sangue (da cui estrarre il DNA) per cominciare ad identificare potenziali fattori genetici collegati alla malattia. Lo ha spiegato Jong-Min Lee, del gruppo di Gusella, che ha individuato due cromosomi candidati. Se identifichiamo nuovi fattori genetici, possiamo pensare di agire su di essi a scopo terapeutico in futuro stimolando l'interesse sempre più presente delle industrie farmaceutiche.
Sviluppo di nuove tecnologie terapeutiche
Parallelamente, la ricerca si avvale di tecnologie sempre più sofisticate ed in grado di interpretare i misteri della malattia sempre di più. È il caso dei modelli di laboratorio oggi disponibili che riproducono aspetti diversi della malattia di Huntington. Una interessante carrellata di cosa oggi esiste nel vasto panorama della ricerca e' stata proposta da Roger Baker di Cambridge, Inghilterra. Oggi disponiamo di un numero impressionante di modelli animali geneticamente modificati che ci consentono di ripercorrere, talvolta, in poche settimane, l'intero decorso della malattia che nell'uomo dura tanti anni. Alcuni di questi modelli sono molto costosi come i primati, mentre altri, come i topi transgenici, possono consentire importanti ricerche con una spesa minore. Questi modelli, purché nel pieno rispetto etico, ci insegnano tantissimo soprattutto per quello che riguarda l'effetto è la tossicità di possibili nuovi farmaci e ci consentono di eseguire studi post mortem, altrimenti impossibili da realizzare nell'uomo. Due sono stati gli studi Italiani presentati su modelli di topo transgenico, entrambi molto promettenti, uno sul ruolo degli astrociti nel metabolismo del colesterolo (gruppo Cattaneo - Università di Milano), l'altro sugli sfingolipidi e sul loro possibile ruolo come target terapeutico (gruppo Squitieri - IRCCS Neuromed ). Ovviamente queste strategie non possono completamente sostituirsi allo studio sull'uomo e mostrano pertanto dei limiti.

Per questo motivo hanno avuto inizio studi importanti come ENROLL-HD, la più grande raccolta di dati e campioni biologici mai esistita prima in una malattia rara. Cosa è e quale è l'obiettivo di ENROLL-HD? Enroll è una piattaforma di ricerca in cui i dati ed i campioni biologici raccolti da pazienti e familiari possono essere condivisi dai ricercatori di ogni parte del mondo. È, in altri termini, un enorme contenitore informatico che costituisce il sistema per favorire la definizione della storia naturale e dell'evoluzione della malattia. Conoscerla in maniera più approfondita ci consentirà di curarla in maniera più approvata. Un esempio è la raccolta dati del Registro Europeo che ha evidenziato il ruolo tossico dell'uso inappropriato di neurolettici sulla severità della progressione della malattia. ulteriori dettagli su questo sito nella sezione ENROLL.
Applicazione delle nuove tecnologie alle sperimentazioni farmacologiche con nuovi composti sviluppati in laboratorio
Nuove tecnologie su applicazione di terapia genica sono attese nel prossimo futuro. A Barcellona sono stati presentati diversi studi che adottano metodologie per contrastare l'effetto tossico della proteina (huntingtina) mutata e tossica. Una riduzione dei livelli di huntingtina mutata nel sistema nervoso dovrebbe non solo proteggere dalla malattia ma anche far regredire alcune delle manifestazioni cliniche già in atto. Questa evidenza è stata acquisita già anni fa da uno studio di un modello di topo transgenico. Tuttavia le metodiche sono complesse, richiedono ancora approfondimenti e, soprattutto, garanzia di tollerabilità. Prima di tutto dobbiamo perciò esser certi che non siano esse stesse tossiche per le persone. Sembrerebbe che la riduzione dei livelli della proteina difettosa, anche se associata ad una riduzione parziale di quella normale, possa rappresentare un rimedio per la malattia. Per questo motivo una prima fase preliminare di studio nei pazienti è prevista nel corso del 2015, come sottolineato da Sarah Tabrizi, nota leader del gruppo Inglese dell'Università di Londra. Siamo di fronte ad una fase, come detto, molto preliminare che necessita di molta prudenza, approfondimento, estremamente sperimentale e da rivolgere, per ora, ad un numero esiguo di pazienti per verificarne innanzitutto la tollerabilità. Le nuove tecnologie dovranno necessariamente tener conto delle differenze genetiche dei pazienti e della loro origine geografica. Ciò è fondamentale per selezionare il gene difettoso senza necessariamente agire su quello sano che è presente, insieme a quello difettoso, in ogni persona con la mutazione. La funzione di quello sano, infatti, è importante ed ancora poco nota. Pertanto un'altra strategia, parallela a quella della terapia genica, è quella di analizzare le caratteristiche del gene Huntington nelle diverse popolazioni. Come si pone l'Italia in questo scenario? La popolazione Italiana risente di influenze genetiche provenienti negli anni da altre popolazioni vicine. Infatti, come emerso dallo studio presentato a Barcellona in collaborazione tra il gruppo di Vancouver (Micheal Hayden), il più esperto al mondo in analisi di questo tipo è quello italiano (Squitieri), la nostra popolazione presenta delle caratteristiche genetiche diverse da quelle del nord Europa. Pertanto anche strategie future di terapia genica dovranno essere adattate a queste diverse caratteristiche. Come vedete la storia è complessa, ma promettente ed è grazie al contributo delle famiglie Italiane e dei campioni biologici che hanno scelto di donare grazie alla mediazione di LIRH che queste informazioni cominciano ad essere ora disponibili.
Quali sono dunque le sperimentazioni attese nel prossimo futuro in Italia e cosa ci dobbiamo aspettare nel breve?

Grosse speranze sono legate alla Pridopidina (ex Huntexil) tuttora in corso. Il punto sulla Pridopidina emerso a Barcellona: Lo studio è complesso per i pazienti e per noi ricercatori. Le valutazioni sono tante, richiedono tempo e spazi, gli appuntamenti da programmare numerosi, il team deve essere quasi del tutto dedicato alla sperimentazione, alcuni dei dosaggi da sperimentare sono più alti di quelli studiati in passato e ciò induce prudenza, più controlli e molta cautela. Per tutti questi motivi il coinvolgimento dei pazienti non è semplice in nessuno dei centri coinvolti. Il centro di Neurogenetica dell’IRCCS Neuromed è stato il primo ad iniziare in Europa ed è attualmente il maggiore reclutatore dello studio. Abbiamo ancora margine per inserire pazienti ed anche gli altri centri italiani dovrebbero a breve iniziare a dare il loro contributo. L'aspettativa sull'efficacia di questo farmaco è altissima e le novità legate alla sua funzione, emerse di recente, promettenti. Speriamo bene. Considerati i ritardi di reclutamento dei centri soprattutto nord americani, è ragionevole attendere risultati non prima di fine 2015/inizio 2016.

La prossima sperimentazione attesa in Italia è quella con Laquinimod (lo studio si chiamerà LEGATO-HD) e coinvolgerà ancora una volta molti paesi del Nord America ed Europa. Per la prima volta in uno studio così allargato sono previste due importanti novità: l'uso della Risonanza Magnetica Nucleare per valutare nel tempo effetti neuroprotettivi ed una condizione anche molto iniziale di malattia, quasi presintomatica. Non possiamo fornire al momento dettagli, che saranno disponibili nei prossimi mesi. Lo studio dovrebbe partire inizio 2015 e siamo pronti a fare la nostra parte. Il farmaco, già in uso nella Sclerosi Multipla, agisce a vari livelli, ma nasce soprattutto come un immunomodulatore per ridurre l'infiammazione tossica documentata nella malattia. Ricorda Fingolimod (FTY720), molecola testata in un modello animale dal gruppo di Neurogenetica diretto dal sottoscritto, di cui avete avuto notizia mesi fa (per approfondimento vedi questo sito). Forse la nostra ricerca sulla neuro protezione nell'Huntington ha contribuito ad aprire un nuovo ambito di ricerca nella terapia della malattia.
Altre sperimentazioni sono in fase di programmazione, ma è forse prematuro farne cenno e speriamo, attraverso un costante intervento di sensibilizzazione di favorire la partecipazione del nostro Paese che non è sempre scontata. In questo senso la frammentazione associativa italiana non aiuta e la considerazione che 'meglio assistere oggi che fare ricerca per aiutare domani' appare incredibilmente miope. In ogni caso mi fa piacere concludere con una ultima notizia positiva che, seppure emersa da internet, ha mantenuto contorni poco chiari e che riguarda la Cisteamina bitartrato, un composto che sembra ridurre la formazione di frammenti tossici della proteina huntingtina mutata, aumenta fattori trofici come il BDNF ed aminoacidi come la cisteina ed avrebbe pertanto proprietà neuroprotettive. Aldilà del nome complicato, lo studio sui pazienti sembra avere avuto risultati promettenti, una preliminare approvazione da autorità regolatorie in attesa di altre in corso ed una reale speranza per il prossimo futuro. Il 2015 rappresenterà, anche per questa storia, un anno di speranza concreta.
A Barcellona si è ovviamente anche parlato di terapie innovative in grado di contrastare direttamente la funzione tossica della proteina huntingtina mutata. In altri termini si è fatto ampiamente cenno alla terapia genica. In particolare lo ha sottolineato la neurologa inglese Sarah Tabrizi che ha fatto cenno all'approccio che si ritiene più atteso nel breve e che, a mio avviso, costituisce una fase ancora preliminare alla terapia genica.
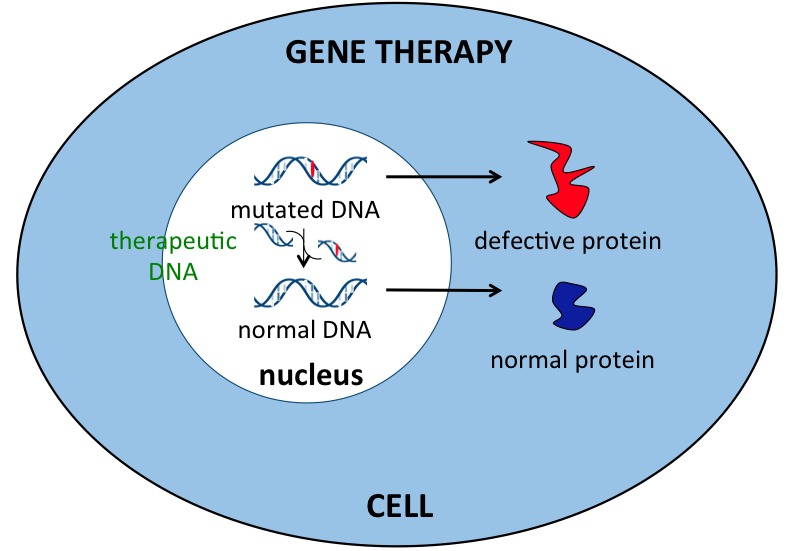
IL PUNTO SULLA TERAPIA GENICA possiamo sensibilmente rallentare il decoroso della malattia o anche far regredire parte dei sintomi. Questi approcci sperimentali sono estremamente promettenti perché agiscono sulla causa alla sua origine. La modalità con cui intervenire può oggi disporre di varie diverse strategie, alcune già tecnicamente realizzabili nell'uomo.
BREVE TERMINE: Una di queste strategie terapeutiche si chiama ASO, allele-specific-oligonucleotide. Cosa significa? È come se noi potessimo disporre di un'arma, una sorta di missile, capace di riconoscere l'arma nemica (l'RNA generato dal gene difettoso) bloccandolo e poi distruggendolo. Se usiamo l'ASO nei nostri modelli in laboratorio abbiamo ottimi risultati. Nell'uomo tuttavia è più difficile perché:
1. L'arma non è precisa e colpisce anche la parte buona del gene Huntington. Nell'animale sembra che questa cosa non sia particolarmente dannosa e anche nei pazienti con un'altra malattia, la SLA familiare (molto più rara della SLA), è ben tollerata anche se non sappiamo ancora se funzioni.
2. Deve essere iniettata direttamente nel liquor cefalorachidiano affinché raggiunga il cervello e raggiunge le sue parti più profonde (quelle anche molto colpite dalla malattia di Huntington) piuttosto poco e male.
3. Non sappiamo quanto a lungo possa agire, quante volte vada ripetuta è che danno nel tempo possa fare. Ricordo che se spegnamo del tutto il gene 'normale' nel topo procuriamo un danno cerebrale grave.
Tuttavia, come Sarah Tabrizi ha sottolineata, una prima sperimentazione di fase 1 per valutare la tossicità (non ancora l'efficacia) della procedura nell'uomo è prevista nel corso 2015. Sarà inizialmente su un numero limitato di pazienti per via dei rischi sopra riportati. È senz'altro un primo importante passo, ma è cosa prudente non sbilanciarsi in questa fase.
Qual'è dunque il futuro di strategie terapeutiche di questo tipo?
MEDIO TERMINE (anni): Utilizzare 'armi' più selettive in grado di bloccare solo l'azione tossica del gene difettoso. Si tratta della cosiddetta 'medicina personalizzata'. Per realizzarla occorre studiare la genetica dei pazienti. È come se ognuno di noi avesse una specie di impronta digitale sul proprio gene Huntington. Per ognuno occorre l'arma giusta che varia nelle popolazioni con origine etnica diversa. Intanto cominciamo oggi a capire che tipo di caratteristiche ha la nostra popolazione in modo da usare domani l'arma giusta. Il contributo delle famiglie è cruciale così come la disponibilità a donare sangue per ricerca per consentirci di esplorare le diverse 'impronte digitali' in ciascuno che ha la mutazione. LIRH e Neuromed collaborano con il gruppo di Vancouver di Hayden che sta esplorando le caratteristiche di tutte le popolazioni del mondo. Le caratteristiche della nostra popolazione, grazie al sangue che ci avete fornito, sono state descritte a Barcellona in uno studio collaborativo tra Italia (gruppo Squitieri) e Canada (gruppo Hayden).
LUNGO TERMINE: possibilità non solo di mirare in maniera corretta, ma anche con i mezzi efficaci a favorire una corretta distribuzione dell'arma letale contro il gene difettoso nei punti giusti del cervello. Un esempio è la terapia genica che fa uso di vettori virali. In questo caso il virus è amico e fa da shuttle al farmaco che blocca l'azione del gene trasportandolo dove è necessario. La difficoltà consiste nell'essere certi che il virus rimanga amico e non si trasformi in un altro nemico capace di far ulteriori danni. Un'altra possibilità è che l'arma non sia solo precisa, ma precisissima ed in grado non solo di individuare il gene mutato ma addirittura solo la mutazione all'interno del gene estirpando le CAG in eccesso. Qualcuno ci sta già provando nell'animale e comincia a riuscirci. È un enorme passo avanti ma ancora un po' distante dalla realizzazione bell'uomo.
Conclusione
Le terapie da cui si attende riscontro nel breve sono quelle farmacologiche (già in fase 2 o 3), molto promettenti nei prossimi due anni (anche se forse non ancora del tutto risolutive) e probabilmente in grado di essere associate tra di loro nel prossimo futuro in modo da prevedere terapie combinate.
La terapia genica è una speranza per il futuro.
Le cellule staminali hanno fatto un prudente passo indietro e i gruppi che autorevolmente se ne sono occupati (in particolare Steve Dunnett ed Anne Rosser in Gran Bretagna), stanno ora riguardandola con un occhio diverso per valutare approcci strategici più adeguati dopo, non si può non sottolineare, il fallimento di questi anni.