
The origin of Huntington Disease in the Middle East described for the very first time: Why this is relevant for possible future treatments

15/07/2020
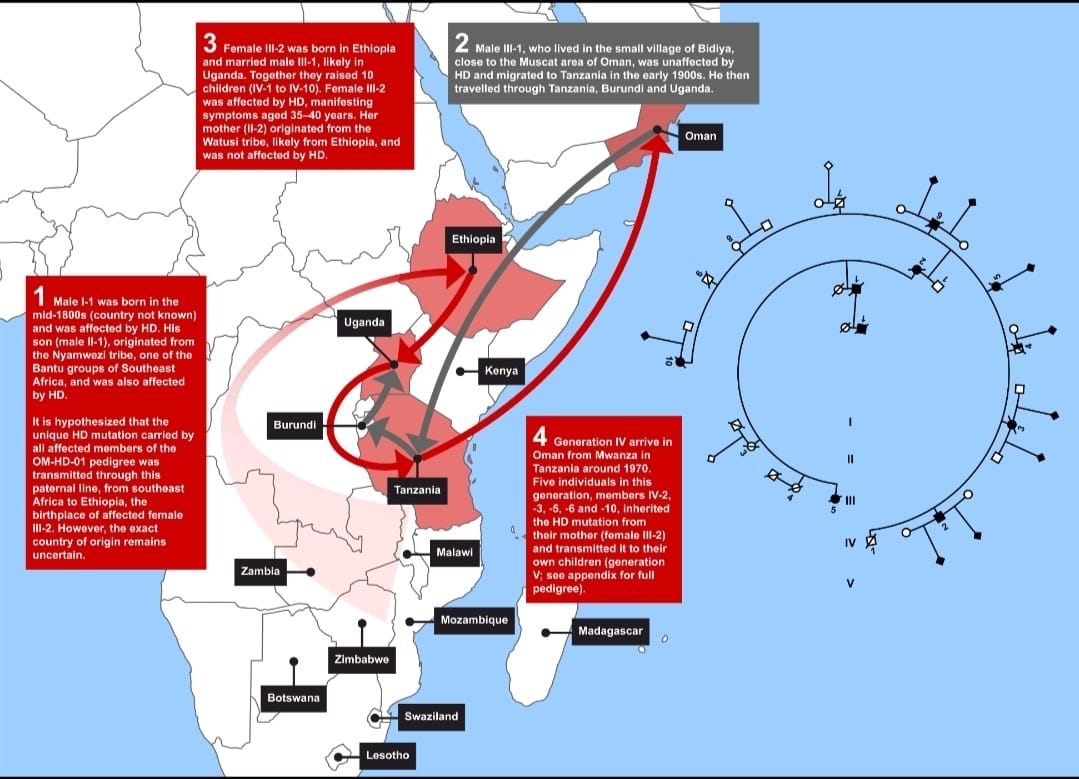
Identified the Mmutated HTT and haplotype of the African ancestor who spread Huntington disease into the Middle East from sub-Saharan Africa , that at the beginning of the 1900s spread Huntington into the Middle East from South Africa,with determining in that area the highest concentration of HD Huntington and juvenile-onset Huntington disease in this part of the world, now identified.
The study, coordinated by Ferdinando Squitieri - just published in the Nature journal "Genetics in Medicine" - the official Journal of the American College of Medical Genetics and Genomics (ACMG), sheds a light on an important missing piece of Huntington 's disease (HD) history and on possible novel treatments of it.
Since 1872, the year in which HD was first described by Dr. George Huntington, the history, etiology and clinical manifestation of the disease named after him has focused mainly on the European and North American populations, also defined Caucasians. In contrast, very little is known about Huntington disease HD in non-European, Black African and Middle East ancestry populations.
Thanks to the genealogical and genetic reconstruction of the largest Middle Eastern sample known to date, it has beenwas possible - for the first time - to determine that Huntington's diseaseHD in a specific area of the Middle East has largely an African and non-European origin, associated with the highest number of juvenile-onset HD (joHD) cases ever described before, including many pediatric-onset patients.
In 2013, the Italian League of Huntington Research Foundation (LIRH) was contacted by a family living in Muscat, the capital of the Sultanate of Oman, desperately searching for information on possible treatments and care for HD, a clinical unknown condition to most of local doctors and institutions. That family reported having been affected by HD over several generations and represents the largest HD cluster (OM-HD-01 pedigree) in Middle East, related to its high number of inter marriages and subsequent heritability risk. “It was an unusual and unexpected experience. When we got the first request-of-help by e-mail from that family, we believed it was like a spam” states Barbara D’Alessio, LIRH Foundation’s Executive Director. “Nevertheless, we did respond to that request and we did our best to help them. That was possible also thanks to the collaboration and sensitivity of the Royal Hospital in Muscat and of the Ministry of Health of the Sultanate”, adds Barbara D’Alessio.
The study was conducted between August 2013 and March 2019 and it focused on a sample of 302 individuals (the OM-HD-01 Pedigree) from the same family. A careful genealogical analysis to determine the ancestral origin of the HD mutation was performed. This information was gathered retrospectively via one-to-one interviews with all surviving members. It has also been carried out a genetic analysis on the DNA from blood samples, which were analyzed in parallel both by the National Genetic Center (NGC), the Ministry of Health Country Hospital in Oman and by the CSS – Mendel Institute in Rome, to determine CAG repeat length in the HTT gene. Single nucleotide-polymorphism (SNP) genotyping was performed at the Center for Molecular Medicine and Therapeutics (CMMT), University of British Columbia, Vancouver (Canada).
The findings confirm that in human history, multiple and rare-disease-causing HTT mutations have arisen spontaneously and independently in different geographical locations, and then moved abroad through migration. As in the ‘800 families from Europe, migrated to North America (in the Boston and New York area, description by George Huntington of 1872) and in South America (area of Venezuela, gene identified thanks to Nancy Wexler's studies in 1993) so, in the same years, HD spread from sub-Saharan Africa into the Middle East. In conclusion, the evidence of a unique HD haplotype originating from South-East Africa, shed the light on a missing piece in HD history.
The population described for the first time shows an earlier onset of the disease , a more aggressive mutation and a greater number of juvenile-onset cases than anywhere else in the world. This conclusion was possible thanks to the comparison of high number of Caucasian patients included in the LIRH database and in ENROLL-HD platform in which LIRH has been participating for years with one of the highest case studies in the world.
These findings lead to significant reflections on the HD knowledge and may have significant impact on future therapies:
- The evidence confirms that in human history several mutations that cause rare diseases have arisen spontaneously and independently in different geographical areas of the world and have then moved abroad via immigration. The question is whether this phenomenon may affect the HD frequency, HD severity and its atypical clinical manifestations (e.g. juvenile-onset disease) worldwide.
- The existence of different genetic characteristics, the so-called haplotypes, that are just like genetic fingerprints, in HD may have a significant impact on the development of a new targeted genetic therapies, such as the allele-specific anti- sense oligonucleotides (ASO). At present, the HTT lowering techniques (improperly defined "gene silencing") target only the most common, predominantly Caucasian, haplotypes. The occurrence of such rare haplotypes in other non-Caucasian populations, suggest that non-selective therapies (either anti-sense or pharmacological) may represent an important resource in the HD treatment.
Once again, it was possible to increase the knowledge on Huntington's diseaseHD and joHD thanks to the contribution of patients, who made available their time and biological samples to research. The same patients founded the Oman's HD Association, which closely collaborates with the Italian Huntington Research Foundation. The study was carried out by an international team coordinated by Ferdinando Squitieri, Head of the Huntington Unit and Rare Diseases Unit of the IRCCS Casa Sollievo della Sofferenza/CSS-Mendel and Scientific Director of the LIRH Foundation and has included researchers from Italy, Germany, Sultanate of Oman, South-Africa and Canada. "It is an historical success that sheds the light on an area of the world that represented, till now, a missing piece for HD history and its research " stated Ferdinando Squitieri, who adds : “We still need to learn more about how genetics affects clinical presentation and severity of HD and joHD”., concludes Ferdinando Squitieri
Read the article here